- C5Neurology
- NMOSD
- 製品情報
- ソリリス® 製品基本情報
- PREVENT試験
第Ⅲ相国際共同臨床試験
[ECU-NMO-301試験(PREVENT試験)]
(多施設共同無作為化プラセボ対照二重盲検比較試験)
視神経脊髄炎スペクトラム障害(NMOSD)患者を対象とした臨床試験は、すべて髄膜炎菌ワクチン接種下で実施されました。
社内資料:第Ⅲ相プラセボ対照二重盲検比較臨床試験(ECU-NMO-301)(承認時評価資料)
Pittock SJ, et al. N Engl J Med 2019; 381: 614-625
[利益相反:本試験はAlexion Pharmaceuticalsの支援のもと実施された]
試験概要
| 目的 | NMOSD患者に対するソリリス®の安全性と有効性を評価する。 |
|---|---|
| 対象 |
抗AQP4抗体陽性のNMOSD患者143例(うち日本人14例):
|
| 方法 | 対象患者をソリリス®群又はプラセボ群に2:1の割合で無作為に割付け、ソリリス® 900 mg又はプラセボを週1回、4週間投与し、その1週間後からソリリス® 1200 mg又はプラセボを2週に1回静脈内投与した。本試験はtime-to-event型試験のため、患者ごとに投与期間は異なる。全ての患者が、試験終了時又は早期中止来院時(再発した患者は、再発後6週時点の追跡来院が試験終了時となる)まで、投与を継続した。 なお、再発又は試験終了のため試験を完了した患者は、非盲検継続試験(ECU-NMO-302試験)に参加できることとした(非盲検継続試験に参加しない患者は8週間の安全性追跡調査期間に移行した)。 |
| 評価項目 |
主要評価項目: 試験中再発:試験中に発現する急性再発発作[24時間超持続する神経学的検査上の客観的変化(臨床徴候)を伴う神経学的症状の新たな発現又は悪化]と定義。徴候及び症状はNMOに起因するもので、関連する臨床的所見を伴わずMRI又はその他の画像検査所見にのみ異常が認められる場合は試験中再発とはみなさない。試験中再発の前30日以上にわたり臨床的に安定していなければならない。 当初の主要評価項目は「試験担当医師により判定された初回の試験中再発までの期間」であったが、施設間でばらつきが認められたことから、治験実施計画書の改訂を行い、主要評価項目を「独立評価委員会により判定された初回の試験中再発までの期間」へ変更した。 主な副次評価項目:
|
| 解析計画 | 有効性及び安全性の解析対象集団は、無作為化されソリリス®又はプラセボを1回以上投与された全患者(FAS)とした。 主要評価項目であるRAC判定による初回の試験中再発までの期間については、無作為割付けの層別化変数を含む層別ログランク検定を用いて投与群間比較を実施し、信頼区間及びp値を示すとともに(有意水準p≦0.05)、ハザード比及び層別化Cox比例ハザードモデルからのリスク低下率を要約した。感度分析として、試験担当医師判定による初回の試験中再発までの期間について、非層別ログランク検定及び多変量Cox比例ハザードモデル(共変量:投与群、中央値で二分したベースラインのEDSSスコア、観察されたIST使用状況及び過去の年間再発率)を用いた解析を事前に規定した。 副次評価項目については、RAC判定による年間再発率は、投与群、過去の年間再発率及び無作為割付けの層別化変数を共変量としたポアソン回帰分析により解析した。試験終了時におけるEDSS、mRS、HAI及びEQ-5D-3Lスコアのベースラインからの変化量は、ベースライン値で調整し無作為割付けの層別化変数によって層別化したノンパラメトリック共分散分析(ANCOVA)を実施した。なお、副次評価項目の解析において、投与群間比較は以下の順位による閉検定手順を用いて実施した:(1)RAC判定による年間再発率、(2)試験終了時におけるEDSSスコアのベースラインからの変化量、(3)試験終了時におけるmRSスコアのベースラインからの変化量、(4)試験終了時におけるHAIスコアのベースラインからの変化量、(5)試験終了時におけるEQ-5D-3Lスコアのベースラインからの変化量。仮説検定は、(1)から(5)へと進め、いずれかの項目が統計学的に有意(有意水準p≦0.05)でなかった場合、それより下位の順位の評価項目は統計学的な検討結果は意味のないものとして扱った。また、次の通り感度分析を実施した:(1)ベースライン値と無作為割付けの層別化変数により調整したANCOVAを用いたベースラインから試験終了時の変化量、(2)共変量としてベースライン値、実際の層別、投与群及び試験来院を組み込んだ反復測定混合モデルを用いた、全ての規定来院時でのベースラインからの変化量、(3)共変量としてベースライン値、実際の層別、投与群、試験来院、及び投与群交互作用別の試験来院を組み込んだ反復測定混合モデルを用いた、1年を超える全ての規定来院時でのベースラインからの変化量。 |
4. 効能又は効果(抜粋)
視神経脊髄炎スペクトラム障害(視神経脊髄炎を含む)の再発予防
5. 効能又は効果に関連する注意(抜粋)
〈視神経脊髄炎スペクトラム障害(視神経脊髄炎を含む)の再発予防〉
5.11 本剤は、抗アクアポリン4抗体陽性の患者に投与すること。
5.12 視神経脊髄炎スペクトラム障害(視神経脊髄炎を含む)※の患者に使用すること。
「多発性硬化症・視神経脊髄炎診療ガイドライン2017」(日本神経学会)を参考にすること。
視神経脊髄炎スペクトラム障害(NMOSD)を対象とした臨床試験で用いた主な評価ツール
| 評価項目 | スコアリング法 | NMO-301試験 | NMO-302試験 | 評価者 |
| 初回の試験中再発までの期間 (RACによる判定) |
試験薬初回投与からRACにより判定された初回の試験中再発までの日数、又は試験中再発が認められなかった場合当該被験者の試験期間の日数 | 主要評価項目 | その他の有効性に関する評価項目 | 医師評価及び RAC評価 |
| 初回の試験中再発までの期間 (試験担当医師による判定) |
試験薬初回投与から試験担当医師により判定された初回の試験中再発までの日数、又は試験中再発が認められなかった場合当該被験者の試験期間の日数 | 主要評価項目の感度分析 | その他の有効性に関する評価項目 | 医師評価 |
| 年間再発率 (RACによる判定) |
RACにより判定された試験中再発の件数を患者・年で割った値 | 副次評価項目 | 主要評価項目の 感度分析 |
医師評価及び RAC評価 |
| 年間再発率 (試験担当医師による判定) |
試験担当医師により判定された試験中再発の件数を患者・年で割った値 | 副次評価項目の感度分析 | 主要評価項目 | 医師評価 |
| EDSS | 0.5刻みで0(神経学的検査正常)から10 (死亡)までの20段階 | 副次評価項目 | 副次評価項目 | EDSS評価者 (盲検下) |
| mRS | 0(まったく症状なし)から6(死亡)の7段階 | 副次評価項目 | 副次評価項目 | 医師評価 |
| HAI | 0(無症候;完全に活動的)から9(常に車椅子を使う;自分自身で移動することができない)の10段階 | 副次評価項目 | 副次評価項目 | 医師評価 |
| EQ-5D-3L | Index:5項目の設問に対して、3段階で回答し、その回答パターンにより、0 (死亡)~1(完全な健康)の基準化された効用値に換算する。VAS:0(想像できる最も良い健康状態)から100(想像できる最も悪い健康状態)までの連続尺度 | 副次評価項目 | 副次評価項目 | 患者による報告 |
:EDSS(Expanded Disability Status Scale;総合障害度評価尺度)は、Kurtzke neurological assessmentに基づく神経障害の指標。標準の神経学的検査の観点から評価された7つの機能系(錐体路機能、小脳機能、脳幹機能、感覚機能、膀胱直腸機能、視覚(眼)機能及び精神機能)のスコアと歩行機能、患者の運動能及び補助器具の使用に関する情報等と組み合わせてEDSSスコアが算出される。
:mRS(Modified Rankin Scale)は、神経障害を有する患者の日常活動の障害度又は依存度の測定に使用される一般的なスケール。
:HAI(Hauser Ambulation Index)は、患者が25フィート(8 m)歩くのに要する時間と支援の程度に基づく神経学的機能(歩行機能)の指標。
:EQ-5D-3L(EuroQoL 5 Dimension 3-Level)は、健康に関する生活の質(QOL)を表し評価する疾患非特異的な手法。移動、身の回りの管理、普段の行動、痛み/不快感及び不安/ふさぎ込みの5項目について「問題なし」、「いくらか問題がある」、「大きな問題がある」の3段階で回答してもらい、基準化された効用値に換算するEQ-5D-3L Indexと、視覚アナログスケールを用いて現在の健康状態の自己評価を捉えるEQ-5D-3L VASで評価された。
有効性
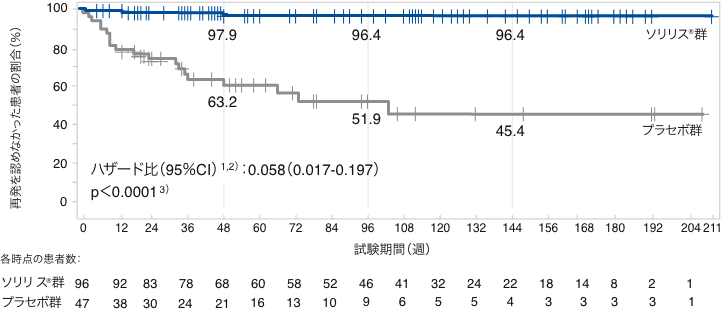
(1)初回の試験中再発までの期間(RAC判定)(主要評価項目、検証的な解析結果)
試験中の初回再発はソリリス®群で3例(3.1%)、プラセボ群で20例(42.6%)に認められ、初回再発までの期間に投与群間で統計学的に有意差が認められました(p<0.0001)(検証的な解析結果)。プラセボに対するソリリス®のハザード比は0.058(95%信頼区間:0.017-0.197)であり、再発リスク(相対リスク)を94.2%低下させました。
初回の試験中再発までの期間(RAC判定、FAS、検証的な解析結果)
1)層別Cox比例ハザードモデルに基づく
2)Wald信頼区間
3)層別ログランク検定に基づく
評価方法
定期的な神経学的検査は少なくとも12週間ごとに実施し、再発が疑われた際には神経学的精密検査を行った。試験中再発とは、試験中に発現する急性再発発作[24時間超持続する神経学的検査上の客観的変化(臨床徴候)を伴う神経学的症状の新たな発現又は悪化]と定義し(徴候及び症状は視神経脊髄炎に起因するもので、関連する臨床的所見を伴わずMRI又はその他の画像検査所見にのみ異常が認められる場合は試験中再発とはみなさない。試験中再発の前30日以上にわたり臨床的に安定していなければならない)、盲検下で再発に関する独立評価委員会(RAC)が判定した。
事前に規定した感度分析である、試験担当医師判定による試験中の初回再発は、ソリリス®群で14例(14.6%)、プラセボ群で29例(61.7%)に認められ、初回再発までの期間に投与群間で統計学的に有意差が認められました(p<0.0001 名目上のp値、層別ログランク検定)。プラセボに対するソリリス®のハザード比は0.180(95%信頼区間:0.095-0.343)で、再発リスクは82.0%低下し、感度分析の結果は、主要解析で得られた結果を支持するものでした。
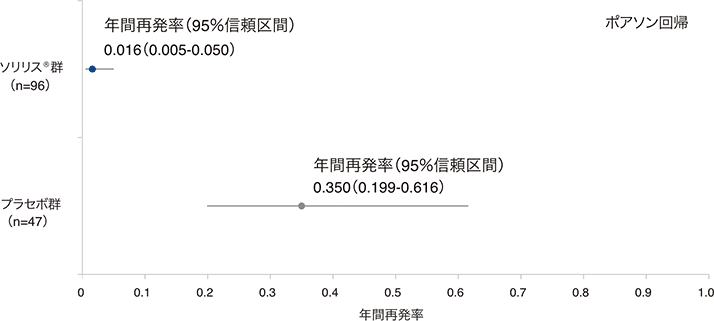
(2)年間再発率(RAC判定)(副次評価項目)
試験中再発の総数はソリリス®群3件及びプラセボ群21件であり*、調整年間再発率はそれぞれ0.016(95%信頼区間:0.005-0.050)及び0.350(95%信頼区間:0.199-0.616)でした。年間再発率のプラセボ群に対するソリリス®群の比は0.045(95%信頼区間:0.013-0.151、p<0.0001、ポアソン回帰※)であり、プラセボ群と比較してソリリス®群で年間再発率が95.5%低下しました。
:再発はソリリス®群の3例で3件、プラセボ群の20例で21件認められました。
年間再発率(RAC判定、FAS)
評価方法
定期的な神経学的検査は少なくとも12週間ごとに実施し、再発が疑われた際には神経学的精密検査を行った。試験中再発とは、試験中に発現する急性再発発作[24時間超持続する神経学的検査上の客観的変化(臨床徴候)を伴う神経学的症状の新たな発現又は悪化]と定義し(徴候及び症状は視神経脊髄炎に起因するもので、関連する臨床的所見を伴わずMRI又はその他の画像検査所見にのみ異常が認められる場合は試験中再発とはみなさない。試験中再発の前30日以上にわたり臨床的に安定していなければならない)、盲検下で再発に関する独立評価委員会(RAC)が判定した。
:投与群、過去の年間再発率(スクリーニング前の24ヵ月間に生じた再発に基づく)及び無作為割付けの層別化変数を共変量とした。
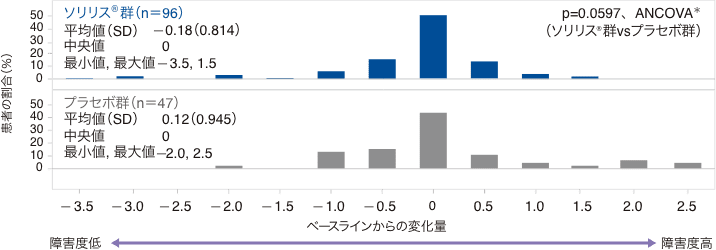
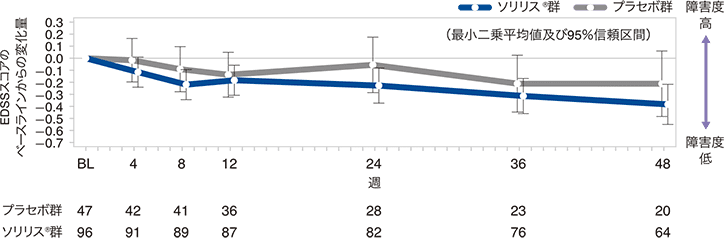
(3)EDSSスコアのベースラインからの変化量(副次評価項目)
試験終了時におけるEDSSスコアのベースラインからの変化量の中央値(最小、最大)は、ソリリス®群で0(−3.5、1.5)、プラセボ群で0(−2.0、2.5)で統計的有意差が認められなかったため(p=0.0597、ANCOVA*)閉検定手順を終了しました。EDSSスコアの変化量が0.5ポイント以上低下した患者の割合はソリリス®群29.2%、プラセボ群29.8%、変化なしの患者がそれぞれ51.0%、42.6%、0.5ポイント以上増加した患者が19.8%、27.7%でした。感度分析の結果は、主要解析の結果と一致していました。
:ベースラインスコアで調整され、無作為割付けの層別化変数(ベースラインのEDSSスコア及びIST実施の有無)によって層別化した、無作為化に基づくノンパラメトリック共変量分散
EDSSスコアのベースラインからの変化量の分布(FAS)
SD:標準偏差
EDSSスコアのベースラインからの変化量の推移:反復測定混合効果モデルによる感度分析(FAS)
(4)mRSスコアのベースラインからの変化量(副次評価項目)
試験終了時におけるmRSスコアのベースラインからの変化量の中央値(最小、最大)は、ソリリス®群で0(−4、2)、プラセボ群で0(−2、3)でした。mRSスコアの変化量が1ポイント以上低下した患者の割合は、ソリリス®群26.0%、プラセボ群10.6%、変化なしの患者がそれぞれ67.7%、74.5%、1ポイント以上増加した患者がそれぞれ6.3%、14.9%でした。
mRSスコアのベースラインからの変化量の分布(FAS)
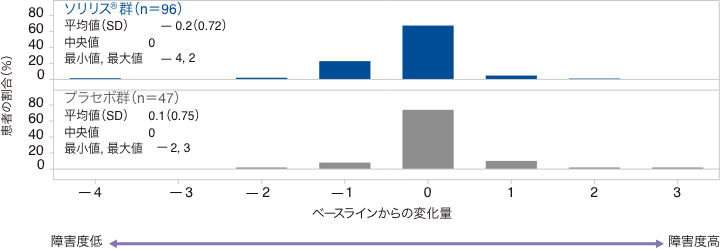
SD:標準偏差
mRSスコアのベースラインからの変化量の推移:反復測定混合効果モデルによる感度分析(FAS)
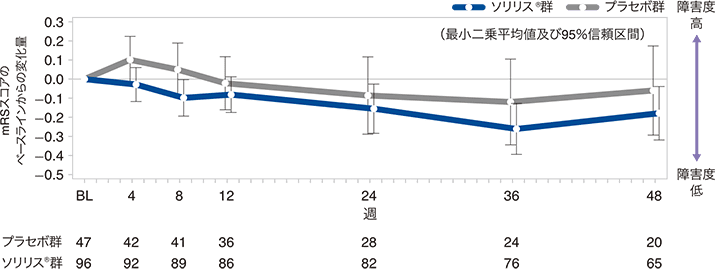
(5)HAIスコアのベースラインからの変化量(副次評価項目)
試験終了時におけるHAIスコアのベースラインからの変化量の中央値(最小、最大)は、ソリリス®群で0(−5、3)、プラセボ群で0(−2、8)でした。HAIスコアの変化量が1ポイント以上低下した患者の割合は、ソリリス®群38.5%、プラセボ群10.6%、変化なしの患者がそれぞれ52.1%、66.0%、1ポイント以上増加した患者が9.4%、23.4%でした。
HAIスコアのベースラインからの変化量の分布(FAS)
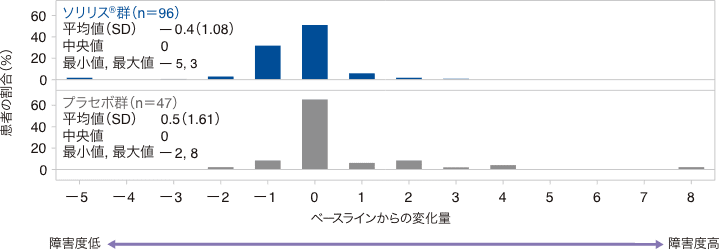
SD:標準偏差
EDSSスコアのベースラインからの変化量の推移:反復測定混合効果モデルによる感度分析(FAS)
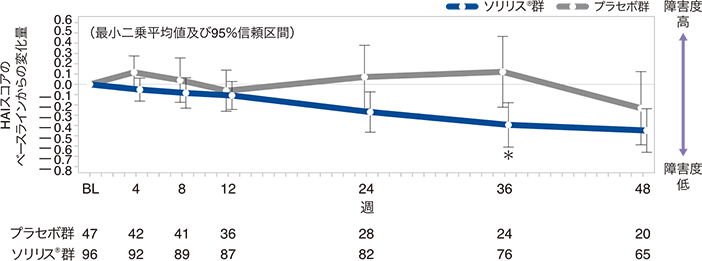
安全性
ソリリス®群の61例(63.5%)及びプラセボ群の34例(72.3%)に副作用が認められました。ソリリス®群で認められた主な副作用は、上気道感染11例(11.5%)、悪心10例(10.4%)、頭痛8例(8.3%)、浮動性めまい7例(7.3%)でした。日本人症例では9例中9例(100.0%)に副作用が認められ、主なものは咽頭炎、膀胱炎、蜂巣炎、ウイルス性胃腸炎の各2例(各22.2%)でした。また、プラセボ群で認められた主な副作用は、頭痛6例(12.8%)、悪心、尿路感染の各5例(各10.6%)、そう痒症4例(8.5%)、咳嗽、嘔吐、肺炎の各3例(各6.4%)でした。日本人症例では5例中3例(60.0%)に副作用が認められ、主なものは頭痛、上咽頭炎、下痢、嘔吐等の各1例(各20.0%)でした。
重篤な有害事象はソリリス®群の30例(31.3%)及びプラセボ群の26例(55.3%)に認められ、視神経脊髄炎スペクトラム障害[ソリリス®群:7例(7.3%)、プラセボ群:16例(34.0%)]、肺炎[ソリリス®群:3例(3.1%)、プラセボ群:1例(2.1%)]等でした。投与中止に至った有害事象は、プラセボ群で2例(肺炎1例、腎前性腎不全及び汎血球減少症1例)に認められました。試験期間中にソリリス®群で1例が副作用(感染性胸水)のために死亡しました。
死亡例も含め、ソリリス®群、プラセボ群において、注目すべき有害事象(AESI)として定義された髄膜炎菌感染症を発現した例はありませんでした。
注)治験薬との因果関係「おそらく関連なし(unlikely)」を含む
