- C5Neurology
- NMOSD
- 製品情報
- ユルトミリス® 製品基本情報
- CHAMPION-NMOSD試験
国際共同第Ⅲ相試験
(補体阻害剤未治療のNMOSD患者を対象とした非盲検外部プラセボ対照試験:CHAMPION-NMOSD試験)1)
1)社内資料:補体阻害剤未治療の視神経脊髄炎スペクトラム障害患者を対象とした国際共同第Ⅲ相試験(CHAMPION-NMOSD試験)(承認時評価資料)
試験概要
| 目的 | [主要目的]補体阻害剤未治療の成人NMOSD患者を対象に「独立評価委員会により判定された試験中再発」に対するユルトミリス®の効果を検証する。 [副次目的]本試験の患者集団におけるユルトミリス®の安全性、年間再発率、疾患関連障害、QOL及び神経機能を評価するとともに、PK/PD及び免疫原性の特性を確認する。 |
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 対象 | 補体阻害剤未治療で抗AQP4抗体陽性の成人(18歳以上)NMOSD患者58例(うち日本人患者は9例)、外部プラセボ対照患者47例(うち日本人患者は5例) | |||||||||||||||||
| 方法 |
第Ⅲ相非盲検外部プラセボ対照多施設国際共同試験。本試験はスクリーニング期(最長6週間)、主要投与期※1、延長投与期で構成された。ユルトミリス®は患者の体重に基づいて、Day1に初回用量、Day15以降は8週ごとに維持用量が点滴静注された。また、外部対照として、NMOSD患者を対象としたエクリズマブの臨床試験(ECU-NMO-301試験)におけるプラセボ群の結果と比較することが事前に規定された。本試験では可能な限り、ECU-NMO-301試験と同様の組み入れ基準、併用治療、評価方法、評価項目が用いられ、試験間の一貫性が保持された※2。
ユルトミリス®投与方法
※3 投与レジメンは記録された直近の試験来院時の体重に基づく。来院日に体重を測定してから投与量を調製するため、通常この体重はその投与時時点のものである。試験薬が来院前夜に調製される場合、直近の試験来院時の体重を使用する。 なお、スクリーニング期に免疫抑制療法(コルチコステロイド、アザチオプリン、ミコフェノール酸モフェチル、メトトレキサート、シクロスポリン及びタクロリムス、シクロホスファミド等)注)が行われていた場合、試験開始後も併用可能とし、NMOSDの再発又は有害事象が発現した場合のみ免疫抑制療法の用量やレジメンの変更を許容した。また、試験中に再発した場合、ステロイドを投与し、必要に応じて血漿交換療法を実施すること及び血漿交換療法や免疫グロブリン静注療法(IVIg)注)を行う場合、試験担当医師の判断でユルトミリス®の補充投与を行うことを可能とした。 注)本邦では一部の免疫抑制療法はNMOSDの適応はない |
|||||||||||||||||
| 主要 評価項目 |
独立評価委員会により判定された初回の試験中再発までの期間(検証的な解析項目) 試験中再発の定義: 試験薬投与期間中に発現した発作で、試験担当医師により判定された24時間以上持続する神経学的検査上の客観的変化(臨床徴候)を伴う神経学的症状の新たな発現又は悪化 |
|||||||||||||||||
| 副次 評価項目 |
独立評価委員会により判定された試験中再発の年間再発率(ARR) |
|||||||||||||||||
| 解析計画 |
有効性の主解析対象集団はFull Analysis Set(FAS)とした。主要評価項目である「独立評価委員会により判定された初回の試験中再発までの期間」のユルトミリス®群と外部プラセボ群との比較では、log-rank検定(両側有意水準5%)を使用し、投与群を因子としたCox比例ハザードモデルによりハザード比及びリスク低下を要約した。「独立評価委員会により判定された試験中再発」が認められなかった場合はFirth法でハザード比、リスク低下、プロファイル尤度95%信頼区間(CI)を推定することとした。また、各評価時点で「独立評価委員会により判定された試験中再発」が認められなかった患者割合の推定値とその95%CIを算出し、両群のKaplan-Meier曲線を示した。なお、ユルトミリス®群と外部プラセボ群との評価期間の均衡を図るため、ユルトミリス®群より追跡期間の長い外部プラセボ群の患者について、ユルトミリス®群での追跡期間の上限までを評価期間とし、それ以降の再発は主解析に含めないこととした。
仮説検定を①から⑥の順序で行い、統計学的に有意でない評価項目(p>0.05)が認められた場合には残りの評価項目は統計学的に有意ではないと判断した。 |
身体障害及びQOLの評価指標
| 評価項目 | スコアリング法 | 報告者 | |
| HAI | 25フィート(約8m)の歩行に要する時間と介助の程度に基づく患者の神経学的機能の測定 | 0(無症候性)〜9(常に車椅子を使用;自分自身で移動することができない) | 医師 |
|---|---|---|---|
| EQ-5D | 健康関連の生活の質(QOL)の測定(EQ-5D indexスコアでは「移動」、「セルフケア」、「普段の活動」、「痛み/不快感」、「不安/抑うつ」の5項目について評価) | Index:5項目の設問に対し「問題なし」、「いくらか問題がある」、「大きな問題がある」の3段階で回答し、その回答パターンにより、0(死亡)~1(完全な健康)の基準化された効用値に換算VAS:0(想像できる最も悪い健康状態)から100(想像できる最も良い健康状態)までの連続尺度 | 患者 |
| EDSS | 錐体路機能、小脳機能、脳幹機能、感覚機能、膀胱直腸機能、視覚機能、精神機能といった中枢神経系の機能及び運動能と日常生活制限の指標による神経症状の測定 | 0.5刻みで0(神経学的検査正常)〜10(死亡) | EDSS 評価者 |
4. 効能又は効果(抜粋)
視神経脊髄炎スペクトラム障害(視神経脊髄炎を含む)の再発予防
5. 効能又は効果に関連する注意(抜粋)
〈視神経脊髄炎スペクトラム障害(視神経脊髄炎を含む)の再発予防〉
5.8 本剤は、視神経脊髄炎スペクトラム障害(視神経脊髄炎を含む)※)の患者に使用すること。
)「多発性硬化症・視神経脊髄炎診療ガイドライン2017」(日本神経学会)を参考にすること。
5.9 抗アクアポリン4抗体陽性の患者に投与すること。
患者背景
| 特性 | ユルトミリス®群 (N=58) |
外部プラセボ群 (N=47) |
|
性別、n(%) 男性 女性 |
6(10.3) 52(89.7) |
5(10.6) 42(89.4) |
|
民族、n(%) ヒスパニック系又はラテン系 ヒスパニック系又はラテン系以外 報告なし 不明 |
9(15.5) 45(77.6) 4(6.9) 0 |
3(6.4) 41(87.2) 1(2.1) 2(4.3) |
|
人種、n(%) アジア人 黒人又はアフリカ系アメリカ人 白人 不明 |
21(36.2) 6(10.3) 29(50.0) 2(3.4) |
15(31.9) 8(17.0) 24(51.1) 0 |
|
初回投与時の年齢(歳) 平均値(標準偏差) 中央値 最小値、最大値 |
47.4(13.84) 46.0 18, 74 |
45.0(13.29) 44.0 21, 75 |
|
初回投与時の年齢分布、n(%) 18歳以上65歳未満 65歳以上 |
51(87.9) 7(12.1) |
44(93.6) 3(6.4) |
|
ベースライン時の体重(kg) 平均値(標準偏差) 中央値 最小値、最大値 |
69.85(19.343) 63.80 41.0, 124.7 |
69.65(16.441) 67.00 46.1, 116.0 |
|
地域、n(%) 米州 欧州 アジア太平洋 |
21(36.2) 17(29.3) 20(34.5) |
15(31.9) 19(40.4) 13(27.7) |
|
ベースライン時の免疫抑制療法注)の使用、n(%) あり ステロイド単独 アザチオプリン アザチオプリン単独 アザチオプリン+ステロイド ミコフェノール酸モフェチル※ ミコフェノール酸モフェチル単独 ミコフェノール酸モフェチル+ステロイド その他の免疫抑制療法 その他の免疫抑制療法単独 その他の免疫抑制療法+ステロイド なし |
28(48.3) 12(20.7) 7(12.1) 3(5.2) 4(6.9) 6(10.3) 2(3.4) 4(6.9) 3(5.2) 2(3.4) 1(1.7) 30(51.7) |
34(72.3) 11(23.4) 13(27.7) 6(12.8) 7(14.9) 8(17.0) 5(10.6) 3(6.4) 2(4.3) 0 2(4.3) 13(27.7) |
|
NMOSD診断時の年齢(歳) 平均値(標準偏差) 中央値 最小値、最大値 |
44.2(14.48) 44.0 17, 73 |
41.1(14.36) 42.0 14, 73 |
|
過去の年間再発率(スクリーニング前24ヵ月間) 平均値(標準偏差) 中央値 最小値、最大値 |
1.87(1.594) 1.44 0.5, 6.9 |
2.07(1.037) 1.92 1.0, 6.4 |
|
再発事象の内訳(スクリーニング前24ヵ月間)、n(%) 視神経炎 片眼性、左眼 片眼性、右眼 両眼性 横断性脊髄炎 一部 全体 一部、縦方向広範囲 全体、縦方向広範囲 脳幹症状 大脳症状 その他の症状 |
25(43.1) 13(22.4) 8(13.8) 6(10.3) 34(58.6) 14(24.1) 8(13.8) 17(29.3) 5(8.6) 9(15.5) 6(10.3) 0 |
22(46.8) 10(21.3) 11(23.4) 5(10.6) 42(89.4) 24(51.1) 4(8.5) 19(40.4) 10(21.3) 15(31.9) 5(10.6) 10(21.3) |
|
ベースラインのHAIスコア 平均値(標準偏差) 中央値 最小値、最大値 |
1.2(1.42) 1.0 0, 7 |
2.1(1.40) 2.0 0, 6 |
|
ベースラインのEQ-5D Indexスコア 平均値(標準偏差) 中央値 最小値、最大値 |
0.766(0.2203) 0.815 0.04, 1.00 |
0.680(0.1961) 0.706 0.27, 1.00 |
|
ベースラインのEQ-5D VASスコア 平均値(標準偏差) 中央値 最小値、最大値 |
73.6(14.81) 77.5 30, 97 |
59.1(20.39) 60.0 0, 95 |
|
ベースラインのEDSSスコア 平均値(標準偏差) 中央値 最小値、最大値 |
3.30(1.584) 3.25 0.0, 7.0 |
4.26(1.510) 4.00 1.0, 6.5 |
本邦適応外
注)一部の免疫抑制療法は本邦未承認であるが、アザチオプリンはNMOSDに保険使用が認められている
本剤には10 mg/mL製剤(ユルトミリス®点滴静注300 mg)と100 mg/mL製剤(ユルトミリス®HI点滴静注300 mg/3 mL及び
ユルトミリス®HI点滴静注1100 mg/11 mL)がありますが、記載のない限り臨床データは10 mg/mL製剤によるものです。
有効性
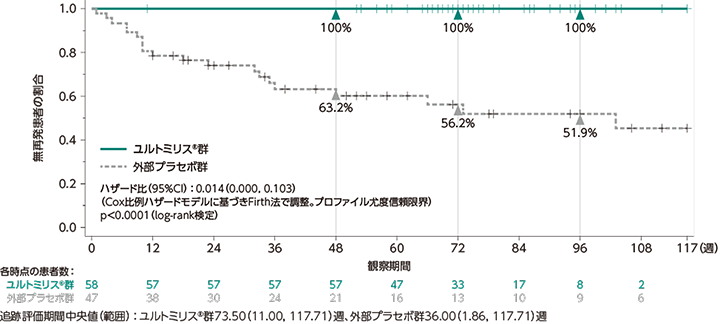
主要評価項目:独立評価委員会により判定された初回の試験中再発までの期間[FAS](サブグループ解析(日本人集団)を含む)
ユルトミリス®群の主要投与期※1における追跡評価期間中央値(範囲)は73.50週(11.00〜117.71週)であり、独立評価委員会により判定された初回の試験中再発は認められませんでした(0例)。一方、外部プラセボ群の追跡評価期間中央値(範囲)は36.00週(1.86〜117.71週)であり、独立評価委員会により判定された初回の試験中再発は20例(42.6%)に認められました。独立評価委員会により判定された初回の試験中再発までの期間について、ユルトミリス®群と外部プラセボ群で統計学的有意差が認められ(p<0.0001※2)(検証的な解析結果)、ユルトミリス®群の外部プラセボ群に対する再発リスクの低下率は98.6%でした[ハザード比(95%CI):0.014(0.000, 0.103)※3]。また、日本人集団におけるユルトミリス®群の追跡評価期間中央値(範囲)は71.86週(53.00〜86.86週)であり、独立評価委員会により判定された初回の試験中再発は認められませんでした(0例)。一方、外部プラセボ群の追跡評価期間中央値(範囲)は34.14週(7.71〜86.86週)であり、独立評価委員会により判定された初回の試験中再発は2例に認められました。
なお、ユルトミリス®群の主要投与期及び延長投与期※4における追跡評価期間中央値(範囲)は90.93週(11.00〜135.14週)であり、独立評価委員会により判定された初回の試験中再発は認められませんでした(0例)。一方、外部プラセボ群の追跡評価期間中央値(範囲)は36.00週(1.86〜135.14週)であり、独立評価委員会により判定された初回の試験中再発は20例(42.6%)に認められました。
- データカットオフ日:2022年3月15日
- log-rank検定
- Cox比例ハザードモデルに基づきFirth法で調整。プロファイル尤度信頼限界
- データカットオフ日:2022年7月15日
独立評価委員会により判定された初回の試験中再発までの期間
(主要評価項目、検証的な解析結果)
独立評価委員会により判定された初回の試験中再発までの期間
(サブグループ解析)
| 日本人集団 | |||
| ユルトミリス®群 (N=9) |
外部プラセボ群 (N=5) |
||
| 独立評価委員会判定で 試験中再発が認められた患者 |
n | 0 | 2 |
| 追跡評価期間(週) | 中央値(最小値、最大値) | 71.86(53.00,86.86) | 34.14(7.71,86.86) |
副次評価項目[FAS]
ユルトミリス®群の主要投与期※1における追跡評価期間中央値(範囲)は73.50週(11.00〜117.71週)でした。一方、外部プラセボ群の追跡評価期間中央値(範囲)は36.00週(1.86〜117.71週)でした。副次評価項目の結果は下表の通りであり、「独立評価委員会により判定された試験中再発のARR」及び「HAIのベースラインからの臨床的に重要な悪化」で統計学的有意差が認められましたが、「EQ-5D indexスコアのベースラインからの変化量」で統計学的有意差が認められなかったため、閉検定手順を終了し、残りの評価項目の統計学的有意差はないと判断しました。副次評価項目の詳細につきましては以降の各項目をご参照ください。
副次評価項目の結果(副次評価項目)
| 副次評価項目 | p値 | |
| 独立評価委員会により判定された試験中再発のARR | <0.0001※2 | |
| HAIのベースラインからの臨床的に重要な悪化 | 0.0228※3 | |
| EQ-5Dのベースラインからの変化量 | EQ-5D indexスコアのベースラインからの変化量 | 0.0567※4 |
| EQ-5D VASスコアのベースラインからの変化量 | − | |
| EDSSのベースラインからの臨床的に重要な悪化 | − | |
- データカットオフ日:2022年3月15日
- 正確確率検定
- ベースラインスコアで調整済みのロジスティック回帰モデル
- 投与群を因子、ベースラインの順位を共変量としたベースラインからの順位付け変化量のANCOVA
副次評価項目:独立評価委員会により判定された試験中再発の年間再発率(ARR)[FAS]
ユルトミリス®群の主要投与期※における追跡評価期間中央値(範囲)は73.50週(11.00〜117.71週)であり、独立評価委員会により判定された試験中再発のARR(95%CI)(副次評価項目)は0.000(NA, 0.044)*でした。事前に比較対照として規定した再発率の0.25(1件/4人年)と比べて統計学的有意差が認められました[p<0.0001(正確確率検定)]。また、日本人集団におけるユルトミリス®群の追跡評価期間中央値(範囲)は71.86週(53.00〜86.86週)であり、独立評価委員会により判定された試験中再発のARR(95%CI)(サブグループ解析)は0.000(NA, NA)でした。
データカットオフ日:2022年3月15日
正確法を用いた95%CIの上限は自由度1のカイ二乗分布に基づき人年数で除した(再発回数が0の場合はCI下限を設定しない)。
独立評価委員会により判定された試験中再発のARR(副次評価項目)

副次評価項目:Hauser Ambulation Index(HAI)のベースラインからの臨床的に重要な悪化[FAS](サブグループ解析(日本人集団)を含む)
歩行機能の評価指標であるHAIについて、ユルトミリス®群の主要投与期※1における追跡評価期間中央値(範囲)は73.50週(11.00〜117.71週)であり、HAIのベースラインからの臨床的に重要な悪化※2は2例(3.4%)に認められました。一方、外部プラセボ群の追跡評価期間中央値(範囲)は36.00週(1.86〜117.71週)であり、HAIのベースラインからの臨床的に重要な悪化は11例(23.4%)に認められました。ユルトミリス®群の外部プラセボ群に対する悪化のオッズ比(95%CI)は0.155(0.031, 0.771)であり、統計学的有意差が認められました[p=0.0228(ロジスティック回帰モデル*)]。また、日本人集団におけるHAIのベースラインからの臨床的に重要な悪化はユルトミリス®群[追跡評価期間中央値(範囲):71.86週(53.00〜86.86週)]及び外部プラセボ群[追跡評価期間中央値(範囲):34.14週(7.71〜86.86週)]ともに認められませんでした。
- データカットオフ日:2022年3月15日
- 臨床的に重要な悪化はベースラインに基づき規定し、「ベースラインが0点、かつ2点以上の増加」又は「ベースラインが0点超、かつ1点以上の増加」とした。
ベースラインスコアで調整済みのロジスティック回帰モデル
HAIのベースラインからの臨床的に重要な悪化(副次評価項目)
| Hauser Ambulation Index(HAI) | 全集団 | ||
| ユルトミリス®群 (N=58) |
外部プラセボ群 (N=47) |
||
| HAIスコアのベースラインからの臨床的に重要な悪化 | n | 58 | 47 |
| なし、n(%) | 56(96.6) | 36(76.6) | |
| あり、n(%) | 2(3.4) | 11(23.4) | |
| 治療効果 | 臨床的に重要な悪化のオッズ比 (ユルトミリス®群/外部プラセボ群) |
0.155 | |
| 95%CI | 0.031, 0.771 | ||
| p値* | 0.0228 | ||
ベースラインスコアで調整済みのロジスティック回帰モデル
HAIのベースラインからの臨床的に重要な悪化(サブグループ解析)
| Hauser Ambulation Index(HAI) | 日本人集団 | ||
| ユルトミリス®群 (N=9) |
外部プラセボ群 (N=5) |
||
| HAIスコアのベースラインからの臨床的に重要な悪化 | n | 9 | 5 |
| なし、n | 9 | 5 | |
| あり、n | 0 | 0 | |
副次評価項目:EQ-5D indexスコアのベースラインからの変化量[FAS](サブグループ解析(日本人集団)を含む)
健康状態(移動、セルフケア、普段の活動、痛み/不快感、不安/抑うつ)を評価したEQ-5D indexスコアについて、ユルトミリス®群の主要投与期※における追跡評価期間中央値(範囲)は73.50週(11.00〜117.71週)であり、EQ-5D indexスコアのベースラインからの変化量の中央値(範囲)は、0.000(−0.33, 0.50)でした。一方、外部プラセボ群の追跡評価期間中央値(範囲)は36.00週(1.86〜117.71週)であり、EQ-5D indexスコアのベースラインからの変化量の中央値(範囲)は0.000(−0.67, 0.41)でした。両群間で統計学的有意差は認められませんでした[p=0.0567(ANCOVA*)]。また、日本人集団におけるEQ-5D indexスコアのベースラインからの変化量の中央値(範囲)は、ユルトミリス®群[追跡評価期間中央値(範囲):71.86週(53.00〜86.86週)]で0.000(−0.10, 0.14)、外部プラセボ群[追跡評価期間中央値(範囲):34.14週(7.71〜86.86週)]で0.000(−0.20, 0.29)でした。
データカットオフ日:2022年3月15日
投与群を因子、ベースラインの順位を共変量としたベースラインからの順位付け変化量のANCOVA
EQ-5D indexスコアのベースラインからの変化量(副次評価項目)
| EQ-5D index スコア | 全集団 | ||
| ユルトミリス®群 (N=58) |
外部プラセボ群 (N=47) |
||
| EQ-5D index スコアのベースラインからの変化量 | n | 58 | 47 |
| 平均値(標準偏差) | 0.005(0.1522) | −0.043(0.2115) | |
| 中央値 | 0.000 | 0.000 | |
| 最小値、最大値 | −0.33, 0.50 | −0.67, 0.41 | |
| p値* | 0.0567 | ||
投与群を因子、ベースラインの順位を共変量としたベースラインからの順位付け変化量のANCOVA
EQ-5D indexスコアのベースラインからの変化量(サブグループ解析)
| EQ-5D index スコア | 日本人集団 | ||
| ユルトミリス®群 (N=9) |
外部プラセボ群 (N=5) |
||
| EQ-5D index スコアのベースラインからの変化量 | n | 9 | 5 |
| 平均値(標準偏差) | 0.004(0.0615) | 0.057(0.1917) | |
| 中央値 | 0.000 | 0.000 | |
| 最小値、最大値 | −0.10, 0.14 | −0.20, 0.29 | |
副次評価項目:EQ-5D VASスコアのベースラインからの変化量[FAS](サブグループ解析(日本人集団)を含む)
患者自身が健康状態を評価したEQ-5D VASについて、ユルトミリス®群の主要投与期※における追跡評価期間中央値(範囲)は73.50週(11.00〜117.71週)であり、EQ-5D VASスコアのベースラインからの変化量の中央値(範囲)は0.5(−45,40)でした。一方、外部プラセボ群の追跡評価期間中央値(範囲)は36.00週(1.86〜117.71週)であり、EQ-5D VASスコアのベースラインからの変化量の中央値(範囲)は0.0(−28, 40)でした。また、日本人集団におけるEQ-5D VASスコアのベースラインからの変化量の中央値(範囲)はユルトミリス®群[追跡評価期間中央値(範囲):71.86週(53.00〜86.86週)]で−1.0(−11, 10)、外部プラセボ群[追跡評価期間中央値(範囲):34.14週(7.71〜86.86週)]で10.0(0, 40)でした。
データカットオフ日:2022年3月15日
EQ-5D VASスコアのベースラインからの変化量(副次評価項目)
| EQ-5D VAS スコア | 全集団 | ||
| ユルトミリス®群 (N=58) |
外部プラセボ群 (N=47) |
||
| EQ-5D VAS スコアのベースラインからの変化量 | n | 58 | 47 |
| 平均値(標準偏差) | 2.6(14.07) | 0.6(16.39) | |
| 中央値 | 0.5 | 0.0 | |
| 最小値、最大値 | −45, 40 | −28, 40 | |
EQ-5D VASスコアのベースラインからの変化量(サブグループ解析)
| EQ-5D VAS スコア | 日本人集団 | ||
| ユルトミリス®群 (N=9) |
外部プラセボ群 (N=5) |
||
| EQ-5D VAS スコアのベースラインからの変化量 | n | 9 | 5 |
| 平均値(標準偏差) | −2.8(7.66) | 14.4(15.63) | |
| 中央値 | −1.0 | 10.0 | |
| 最小値、最大値 | −11, 10 | 0, 40 | |
副次評価項目:EDSSのベースラインからの臨床的に重要な悪化[FAS](サブグループ解析(日本人集団)を含む)
神経症状の評価指標(中枢神経系の機能及び運動能と日常生活制限の指標)のEDSSについて、ユルトミリス®群の主要投与期※1における追跡評価期間中央値(範囲)は73.50週(11.00〜117.71週)であり、EDSSのベースラインからの臨床的に重要な悪化※2は6例(10.3%)に認められました。一方、外部プラセボ群の追跡評価期間中央値(範囲)は36.00週(1.86〜117.71週)であり、EDSSのベースラインからの臨床的に重要な悪化は11例(23.4%)に認められました。また、日本人集団におけるEDSSのベースラインからの臨床的に重要な悪化は、ユルトミリス®群[追跡評価期間中央値(範囲):71.86週(53.00〜86.86週)]で1例に認められ、外部プラセボ群[追跡評価期間中央値(範囲):34.14週(7.71〜86.86週)]では認められませんでした。
- データカットオフ日:2022年3月15日
- 臨床的に重要な悪化はベースラインに基づき規定し、「ベースラインが0、かつ2点以上増加」、「ベースラインが1~5、かつ1点以上増加」、「ベースラインが5を超えており、かつ0.5以上の増加」とした。
EDSSのベースラインからの臨床的に重要な悪化(副次評価項目)
| EDSS スコア | 全集団 | ||
| ユルトミリス®群 (N=58) |
外部プラセボ群 (N=47) |
||
| EDSSのベースラインからの臨床的に重要な悪化 | n | 58 | 47 |
| なし、n(%) | 52(89.7) | 36(76.6) | |
| あり、n(%) | 6(10.3) | 11(23.4) | |
| 治療効果 | オッズ比(ユルトミリス®群/外部プラセボ群) | 0.332 | |
EDSSのベースラインからの臨床的に重要な悪化(サブグループ解析)
| EDSS スコア | 日本人集団 | ||
| ユルトミリス®群 (N=9) |
外部プラセボ群 (N=5) |
||
| EDSSのベースラインからの臨床的に重要な悪化 | n | 9 | 5 |
| なし、n | 8 | 5 | |
| あり、n | 1 | 0 | |
主要評価項目に関するサブグループ解析[FAS]
ユルトミリス®群の主要投与期※における追跡評価期間中央値(範囲)は73.50週(11.00〜117.71週)でした。一方、外部プラセボ群の追跡評価期間中央値(範囲)は36.00週(1.86〜117.71週)でした。主要評価項目の独立評価委員会により判定された初回の試験中再発までの期間についてサブグループ解析を実施したところ、黒人又はアフリカ系アメリカ人を除くサブグループにおいて、ハザード比の95%CIの上限は1未満でした。
データカットオフ日:2022年3月15日
独立評価委員会により判定された初回の試験中再発までの期間に関するサブグループ解析
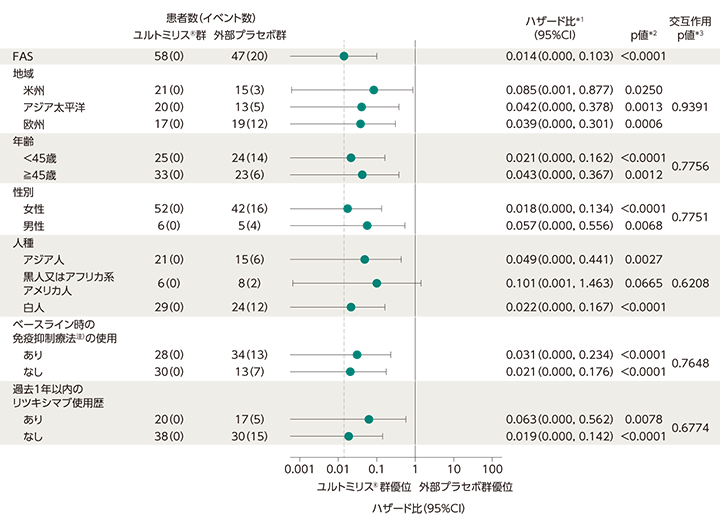
- 投与群を共変量としたCox比例ハザードモデル(プロファイル尤度信頼限界を適用したFirth法で調整)
- log-rank検定
- 交互作用項を含むCox比例ハザードモデル
注)本邦では一部の免疫抑制療法はNMOSDの適応はない
本剤には10mg/mL製剤(ユルトミリス®点滴静注300mg)と100mg/mL製剤(ユルトミリス®HI点滴静注300mg/3mL及びユルトミリス®HI点滴静注1100mg/11mL)がありますが、記載のない限り臨床データは10mg/mL製剤によるものです。
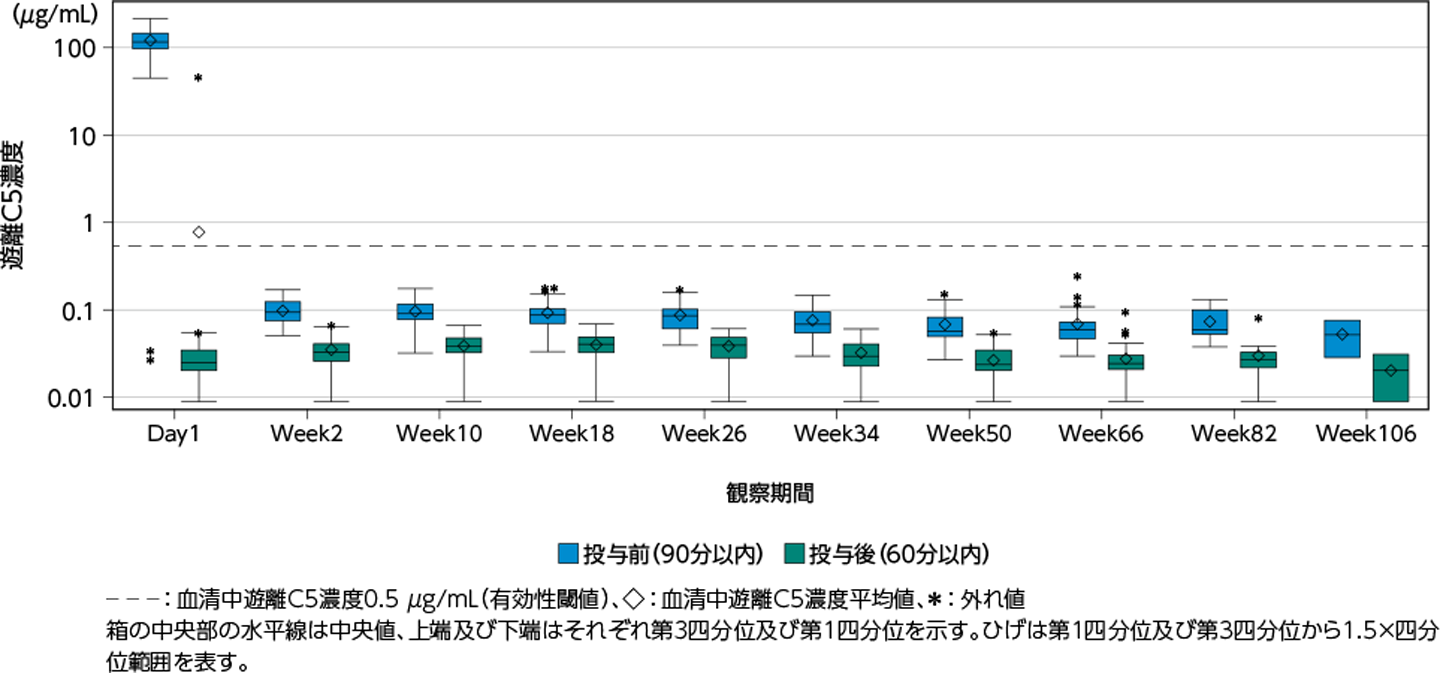
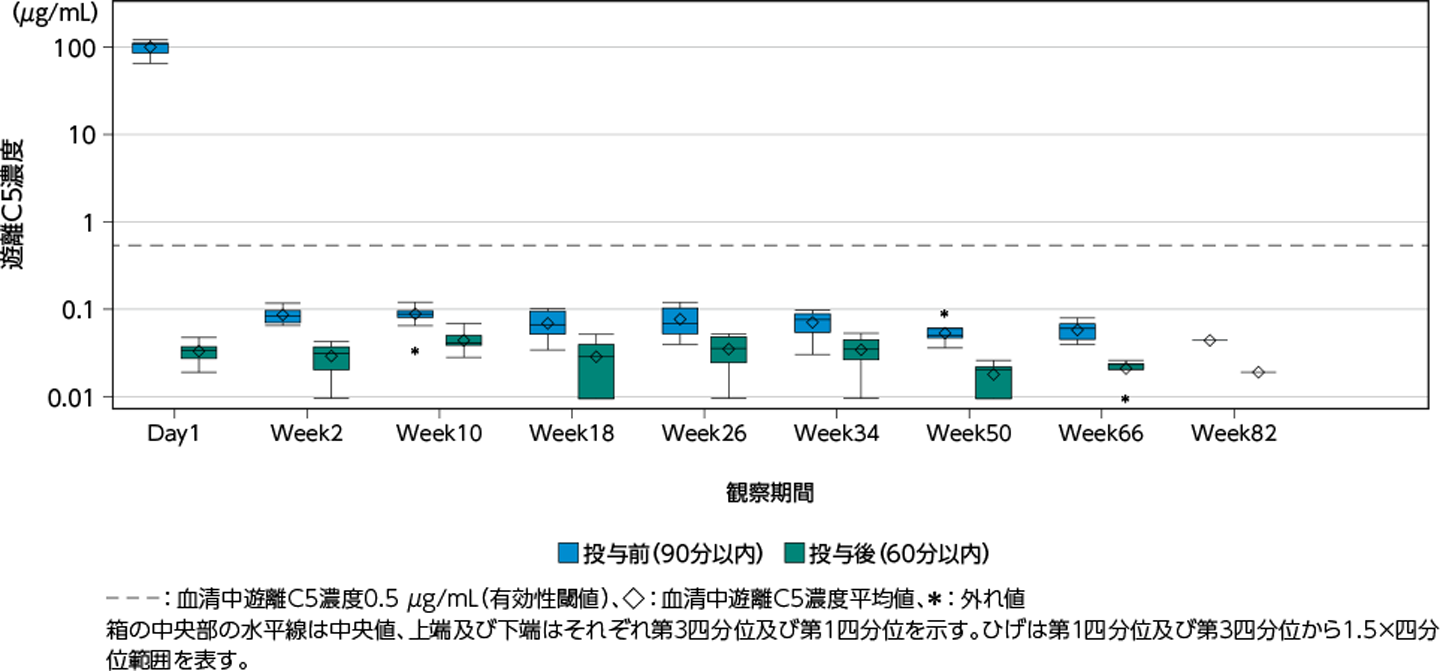
薬力学的効果
ユルトミリス®投与開始後の血清中遊離C5濃度は、試験期間を通して、全集団及び日本人集団ともに有効性の閾値である0.5 μg/mL未満を示しました[外国人集団における1例(Day1投与後)を除く]。
全集団の血清中遊離C5濃度-時間プロファイル
日本人集団の血清中遊離C5濃度-時間プロファイル
本剤には10mg/mL製剤(ユルトミリス®点滴静注300mg)と100mg/mL製剤(ユルトミリス®HI点滴静注300mg/3mL及びユルトミリス®HI点滴静注1100mg/11mL)がありますが、記載のない限り臨床データは10mg/mL製剤によるものです。
安全性
ユルトミリス®投与例は58例(うち日本人は9例)であり、これらを安全性解析対象集団として、主要投与期及び延長投与期※における安全性を評価しました。投与期間中央値(範囲)は89.93週(2.0~131.1週)[日本人集団で82.14週(50.1~98.1週)]でした。副作用は58例中26例(44.8%)に認められ、主な副作用は注入に伴う反応4例(6.9%)及び頭痛3例(5.2%)でした。また、日本人集団における副作用は9例中5例(55.6%)に認められ、その内訳はリンパ球数減少2例(22.2%)、膀胱炎、上気道感染、帯状疱疹、頭痛、紅斑各1例(11.1%)でした。
重篤な有害事象は58例中11例(19.0%)に認められ、その内訳は髄膜炎菌性脳炎、感染、椎間板炎、髄膜炎菌性敗血症、肺炎、足関節部骨折、変形性脊椎症、乳腺浸潤性小葉癌、視神経脊髄炎偽再発、自殺念慮、肺塞栓症、深部静脈血栓症各1例でした。投与中止に至った有害事象は1例(気管支炎、髄膜炎菌性脳炎、ステノトロフォモナス感染)に認められました。本試験において有害事象による死亡は認められませんでした。
データカットオフ日:2022年7月15日
全集団及び日本人集団の有害事象発現状況の概要(安全性解析対象集団)
n(%)
| 全集団 (N=58) |
日本人集団 (N=9) |
|
| 有害事象 | 54(93.1) | 9(100.0) |
|---|---|---|
| 副作用 | 26(44.8) | 5(55.6) |
| 重篤な有害事象 | 11(19.0) | 3(33.3) |
| 投与中止に至った有害事象 | 1(1.7) | 0 |
| 死亡 | 0 | 0 |
全集団で5%以上に発現した有害事象(安全性解析対象集団)
n(%)
| 有害事象 | 全集団 (N=58) |
日本人集団 (N=9) |
| 有害事象 | 54(93.1) | 9(100.0) |
|---|---|---|
| COVID-19 | 20(34.5) | 0 |
| 頭痛 | 18(31.0) | 3(33.3) |
| 関節痛 | 7(12.1) | 1(11.1) |
| 背部痛 | 7(12.1) | 2(22.2) |
| 上気道感染 | 6(10.3) | 2(22.2) |
| 尿路感染 | 6(10.3) | 0 |
| 膀胱炎 | 5(8.6) | 2(22.2) |
| 発熱 | 5(8.6) | 0 |
| 便秘 | 4(6.9) | 0 |
| 浮動性めまい | 4(6.9) | 0 |
| 疲労 | 4(6.9) | 0 |
| 注入に伴う反応 | 4(6.9) | 0 |
| 上咽頭炎 | 4(6.9) | 0 |
| 口腔ヘルペス | 4(6.9) | 1(11.1) |
| 嘔吐 | 4(6.9) | 0 |
| 悪寒 | 3(5.2) | 0 |
| 咳嗽 | 3(5.2) | 0 |
| 下痢 | 3(5.2) | 1(11.1) |
| 排尿困難 | 3(5.2) | 1(11.1) |
| 胃食道逆流性疾患 | 3(5.2) | 0 |
| 高血圧 | 3(5.2) | 0 |
| リンパ節症 | 3(5.2) | 0 |
| 倦怠感 | 3(5.2) | 0 |
| 片頭痛 | 3(5.2) | 0 |
| 筋肉痛 | 3(5.2) | 0 |
| 非心臓性胸痛 | 3(5.2) | 0 |
| 副鼻腔炎 | 3(5.2) | 0 |
| ワクチン接種部位疼痛 | 3(5.2) | 0 |
| 回転性めまい | 3(5.2) | 0 |
注:同一事象が同一患者に複数回発現した場合は1件として取り扱った。有害事象名はMedDRA/J ver. 25.0を用いて表示した。
本剤には10mg/mL製剤(ユルトミリス®点滴静注300mg)と100mg/mL製剤(ユルトミリス®HI点滴静注300mg/3mL及びユルトミリス®HI点滴静注1100mg/11mL)がありますが、記載のない限り臨床データは10mg/mL製剤によるものです。
